Už víme, že naše HLA jsou naprosté děvky. Tak si v tom bordelu pojďme udělat pořádek.
V jednom z předchozích blogpostů takřka před rokem jsem se věnoval HLA molekulám, co jsou zač, konceptu jejich promiskuity a souvislosti s rakovinou. V závěru jsem učinil slib, že se budu taky věnovat bioinformatickým nástrojům na jejich genotyping a další bioinformatickou analýzu. Pokud chcete, osvěžte si nejprve úvod do děvkologie.
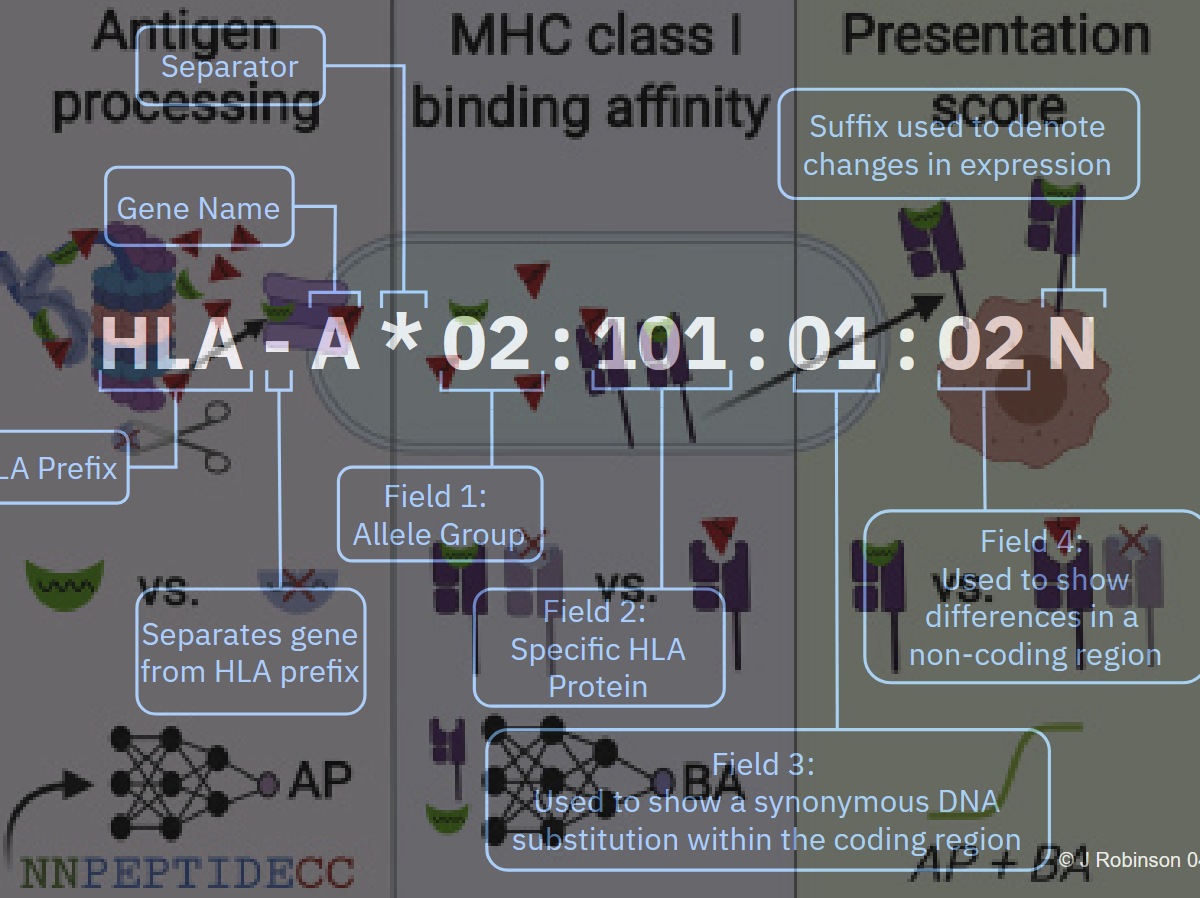
Co je to tedy genotyping HLA a proč se mu věnovat? Genotypingem HLA zjistíme, kterou/é konkrétní alelu/y jedinec má. Výsledek takového zjištění pak vypadá třeba takto: A*02:11:01:01. Nebo taky klidně jenom A*02:11, podle toho co přesně potřebujete. První písmeno značí, o který HLA gen se jedná, první dvojčíslí značí alelickou skupinu, druhé číslo specifickou proteinovou sekvenci, třetí číslo označení různých sekvencí DNA, které jsou ale pro proteinovou sekvenci synonymní (vzniká stejná proteinová sekvence), čtvrté číslo pak rozdíly sekvence v nekódujících oblastech. Zpravidla si vystačíme s prvními dvěma čísly za hvězdičkou.
Na co je dobré mít ogenotypované HLA lokusy? Nejlépe ustálené je využití téhle informace u transplantací: je dobré mít dobře podložený match mezi donorem a recipientem. Z hlediska onkogenetiky je možné najít nějaké asociace mezi přítomností konkrétní alely některého HLA lokusu a rizikem vzniku rakoviny nebo konkrétním typem rakoviny. Znalost konkrétních HLA genotypů se ukazuje jako faktor i u volby imunoterapie, zde už ale často potřebujeme vědět víc než jen genotyp. Čímž se dostáváme na křehký led imunoterapie a personalizované medicíny, možná i neoantigenních vakcín, takže zatím radši rychle zpět.
Pro genotyping potřebujeme mít sekvenační data. Já měl k dispozici data jednak z panelové sekvenace (jde o sekvenaci jen sady vybraných genů) a také tzv. whole exome sequencing data (tj. sekvenaci úseků genomu, které kódují proteiny). Ty mohou existovat buď ve formě hrubých dat (FASTQ, v podstatě jen získané sekvence oštěmplované kvalitou readů), nebo BAM (binary alignment map, ty už prošly alignmentem proti sekvenci referenčního genomu). Protože u BAMů je vždy otázka, co přesně je potřeba správně udělat pro alignment, přišlo mi logičtější používat FASTQ. Porovnával jsem genotypovací nástroje jménem T1K, SpecHLA a HLA-HD. Všechny akceptují FASTQ i BAMy, a nakládají s nimi podle svého. Všechny tyto nástroje běží v C++/C a mají nějakou nadstavbu v Pythonu a obsahují specializovaný postup pro alignment, který je u HLA potřeba kvůli jejich vysokému polymorfismu. Ukázalo se, že T1K a HLA-HD vyprodukují při genotypingu tentýž výsledek, s tím rozdílem, že HLA-HD je schopný vyprodukovat genotyp s rozlišením na čtyři pozice, což ale nikoho nezajímá. Na druhou stranu, T1K produkuje i genotypy KIR a tak nějak celkově působí nejzavedenějším dojmem a jako hotový udržovaný nástroj příjemný na použití. SpecHLA se kupodivu ve svých genotyping výsledcích poněkud odchyloval, ale kromě nich uváděl i “G-group”, což je další nomenklatura HLA alel, která je sdružuje na základě identických nukleotidových sekvencí částí kódujících jejich domény vázající peptidy. Dvě různé HLA alely sdružené pod jednu G-group tedy z pohledu imunitního systému jsou funkčně identické. SpecHLA se v označení těchto G-groups překvapivě shodoval s výsledky T1K a HLA-HD, což tedy nevím přesně jak interpretovat a je to zvláštní.
Ve chvíli kdy máme k dispozici výsledky HLA genotypování se můžeme vrhnout na další analýzy, které vycházejí z těchto nově vygenerovaných datasetů. Jednou z metrik která nás může zajímat je kupříkladu HED skóre. HED = HLA Evolutionary Divergence. Počítá se zde tzv. Granthamova vzdálenost mezi alelami v určitém lokusu, což je hodnota která vychází z fyzikálně-chemické odlišnosti mezi aminokyselinami v peptidu vázaném danou HLA molekulou. Větší diverzita v repertoáru vázaných peptidů, definovaná dvěma HLA alelami v daném lokusu pak znamená větší HED skóre pro daný lokus. Pokud je někdo takový nešťastník, že je v daném HLA lokusu homozygotem (tj. jeho dvě alely jsou totožné, byť alespoň funkčně), má HED skóre rovno nule. Z hlediska onkogenetiky je tento údaj zajímavý jako predikční faktor pro odpověď na imunoterapii, pacienti s vysokým HED skóre mají totiž vyšší šanci na tento typ léčby reagovat, a naopak. Pro výpočet HED skóre existuje jakýsi Perl skript vycházející z původní metodiky výpočtu z roku 1974, já ale používal novější pythonový HLA-HED, tomu ale schází výpočet pro HLA II. třídy, a alespoň některé umí spočítat cHED, dostupný také jako webová aplikace.
Další zajímavá informace je stanovení populační frekvence haplotypů v sadě genotypovaných jedinců. Vlastně, popravdě jsem původně myslel, že najdu nástroj pro stanovení haplotypů, ale ukázalo se, že něco takového ze sekvenačních dat s poměrně krátkými ready, které jsou navíc čistě exomového původu, není prakticky možné. Nástroj Hapl-o-mat je však schopný něco takového odvodit z genotypizačních dat alespoň na úrovni populačních dat. Díky nim je možné se ujistit, zda je v populaci sekvenovaných jedinců standardní rozložení běžných haplotypů, či zda se zde nachází někdo, kdo je potenciálně zajímavý.
Pro účely mimo jiné taktéž haplotypingu jsem zkoušel rovněž nástroj jménem Locityper v kombinaci s Immuannot, a zde jsem dostal zatím strašně přes držku, poněvadž jsem se srazil s pro mě zatím krajně podivným konceptem pangenomů. Totiž, použitím těchto nástrojů jsem nezískal samotný kýžený výsledek, jak bych byl obvykle zvyklý, ale místo toho… označení top pangenomů… které jsou na X % podobné s daným vzorkem. Co přesně pak v daném pangenomu je přítomno za genotyp/haplotyp si pak musím zjistit sám. Whoa. Uvidíme, zda se to ujme, je to poměrně nový koncept, a mně se zatím nelíbí.
Poslední téma, kterému bych se rád věnoval, je moment čiré fascinace, který ve mně uzrál po zprovoznění nástroje s rozkošným názvem MHCflurry. Tento pythonový nástroj má za cíl stanovit metriku zvanou peptide-binding affinity, o které jsem zjistil že je mnohem magičtější, než by se dalo čekat. Do nástroje se vkládají dvě informace: sekvence peptidů (můžou to v kontextu onkogenetiky být například známé sekvence nádorových neoantigenů), a genotypy osekvenovaných jedinců. Vylézají z něj číselná skóre vazebné afinity vložených peptidů, jejich “presentation score”, které vychází z jakýchsi predikcí dalšího procesování daných peptidů. Co mi na tom připadá magické je velký pocitový posun mezi suchými informacemi, jakými jsou jednak genotypy HLA lokusů a jednak peptidová sekvence, a na druhou stranu něco velmi reálného, jako mající “měřitelnou” schopnost vázat tyto peptidické sekvence. To už není jen tabulka čísel, ale kus reálného světa imunologických souvislostí a důsledků, něco velmi uceleného a popisného. Reálné využití této informace je predikce schopnosti imunitního systému vázat nádorové neoantigeny.
Co mě na popisovaných nástrojích trochu děsí je, že čím víc je poznávám, tím častěji zjišťuji, že jsem se v chápání toho co ukazují poněkud mýlíl. Například u SpecHLA jsem se jednu dobu domníval, že dovede detekovat jev typický pro nádorová onemocnění, zvaný Loss of Heterozygosity (LOH) - ztrátu heterozygozyty. Nicméně, s daty, která mám k dispozici to ve skutečnosti nejde, už jen proto, že vycházejí ze sekvenace DNA z periferní krve, kdežto ztráta heterozygozity je jev, který se odehrává v samotném nádoru a vyžaduje pro potvrzení srovnání nádorové tkáně s normální. Podobně strastiplnou cestu jsem měl při snaze o analogické metriky u KIR lokusů, a krom toho, co je to vlastně KIR…? O tom možná někdy jindy.